Table of contents
1. General information
Targeted inhibition of gene expression is a powerful approach in both experimental research aimed at understanding the gene function and therapeutic applications. RNA interference (RNAi) technology provides a diverse set of tools for post-transcriptional gene silencing, among these, artificial miRNA (amiRNAs) are the most complex, consisting of a target-specific siRNA insert embedded within a natural primary miRNA (pri-miRNA) scaffold. In cells, amiRNAs undergo two-step processing by the Drosha and Dicer endonucleases, ensuring their expression levels remain close to endogenous miRNAs. This unique property makes amiRNAs promising therapeutic candidates, combining long-term expression, specificity, and safety with high efficacy.
Here, we present miRarchitect, a web server for the customized design of human amiRNAs. Given the crucial role of sequence and structure in amiRNA processing and activity, efficient computational support is essential for their rational design. A unique advantage of this tool is its use of machine learning to optimize target sequence selection, siRNA insert design, and pri-miRNA scaffold choice, ensuring that the resulting amiRNAs closely resemble endogenous pri-miRNAs in both sequence and structure. The predictive accuracy of miRarchitect was validated in cell culture experiments and demonstrates high predictive performance.
The general idea of miRarchitect is based on the processing path shown in the figure below.
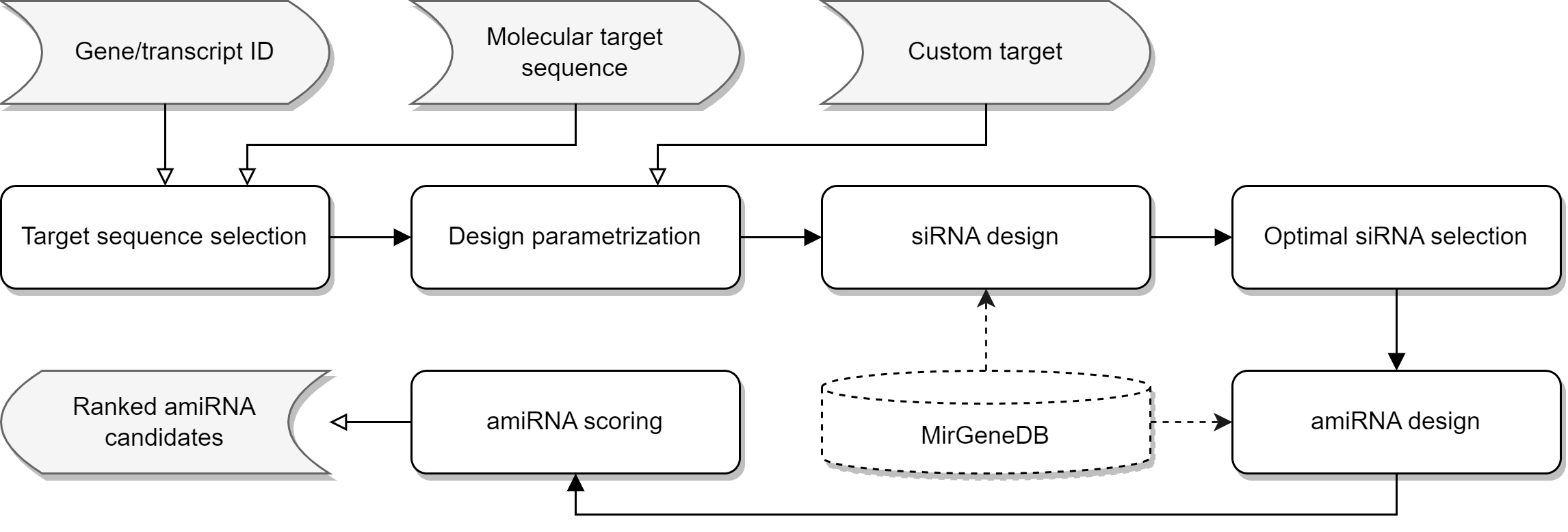
Preview
2. Running miRarchitect
To design an amiRNA using miRarchitect, users provide their data in a 2- or 3-step process. First, they upload the basic data on the Data upload page. If needed, they can further specify these data on the Target specification page before proceeding to configure design parameters.
2.1. Uploading basic data
miRarchitect allows users to work with RNA and DNA sequences by providing input in multiple ways. Users must enter data on this page (named Data upload) to proceed. Once data is provided, the "Proceed" button becomes enabled, allowing users to move to the next steps, where they can complete input specification (if needed) and configure design parameters. The system offers the following options for data input:
- Enter a gene/transcript identifier - Users can input a single gene or transcript identifier (an accession number or name). The system will query the Ensembl database to retrieve the corresponding nucleotide sequence.
- Paste molecular target sequence or insert a custom target sequence - Users can enter sequence in one of two fields. The first field is for a full nucleotide sequence of molecular target, which can range from 21 to 5000 nucleotides in length. The second field allows for a custom target sequence, between 21 and 24 nucleotides long. This option is recommended only when no candidate amiRNA is returned for the full-length molecular target. The short sequence should correspond to a difficult target region. When this option is used, miRarchitect skips the design and target evaluation steps, and the results may include a high number of off-targets. In both fields, sequences must follow on-letter encoding, meaning only A, C, G, U, T are accepted. Fields can be cleared by clicking “x” that appears in the right corner.
- Upload data from a file or use ready-made example - Users can upload a local FASTA file (.fasta) or select a ready-made example to test the system. Six examples are available: examples 1-3 provide a gene/transcript identifier (autofilling option 1), example 4 provides a molecular target sequence (autofilling option 2, first field), and examples 5-6 provide custom target sequences (autofilling option 2, second field).
Additionally, users can retrieve results from a previously processed task directly from the homepage by selecting option:
- Retrieve results from a previous task - Users can enter a task ID to access results of previously submitted job. If the ID is valid and the results are still available, the "Get result" button becomes active, allowing users to view the stored output.
2.2. Specifying the target
The Target specification page appears only if the user provided a gene/transcript identifier (accession number or name) in the Data upload step. This also applies when selecting example 1, 2, or 3. If the input was a full nucleotide sequence or custom target sequence, this step is skipped, and users proceed directly to the Design parameterization page.
On the Target specification page, users first see a list of genes and/or transcripts associated with the identifier they provided. For example, if the user entered NM_005656 (or selected example 1), the displayed list includes a gene with ID ENSG00000184012 and a transcript with ID ENST00000332149. From this list, users should select the target of interest by clicking the appropriate radio button on the left. After making a selection, a transcript specification table appears below. This table either displays the chosen transcript (if a transcript was selected in the first table) or a list of transcripts associated with the gene (if a gene was selected in the first table). For each transcript in the second table, users can view its detailed specification, including ID, start and end positions, length, name, number of exons, and version. By default, the first transcript in the list is pre-selected; however, users can change this selection if the table contains more than one transcript.
Both tables on the Target specification page are displayed in subtables of 10 entries per page if they contain more than ten elements. Stable identifiers of genes and/or transcripts listed in these tables are active links to the Ensembl database. When users click on any identifier, they are directed to the respective page in Ensembl for more detailed information.
After making the selection(s) on this page, the "Proceed" button becomes active, allowing the users to continue to the Design parameterization step. Users can also return to the Data upload page by clicking "Go back". If they do so after selecting a transcript, its sequence will automatically appear in the input field, enabling users to modify it before starting the design procedure.
2.3. Setting design parameters
The Design parameterization page is the final one to visit before running the amiRNA design process. It allows users to select a pri-miRNA scaffold from five available options and customize the design parameters as needed.
miRarchitect supports five human endogenous pri-miRNAs as scaffolds: pri-miR-21, -30a, -122, -135b, and -155. These scaffolds were selected based on their well-characterized and precise cleavage by Drosha and Dicer, as validated in miRGeneDB. By default, four of them (pri-miR-21, -30a, -122, and -155) are pre-selected. The pri-miR-135b scaffold, which may produce mature miRNAs from both arms, is not selected by default, as such dual-arm processing is not recommended for therapeutic applications.
| Pri-miRNA scaffold | hsa-mir-21 | hsa-mir-30a | hsa-mir-122 | hsa-mir-135b | hsa-mir-155 |
|---|---|---|---|---|---|
| scaffold sequence length | 120 nt | 123 nt | 118 nt | 121 nt | 121 nt |
| selected by default? | yes | yes | yes | no | yes |
The list of design parameters includes 11 configurable options. By default, all of them are pre-set to values recommended by the miRarchitect's team, and the suggested pri-miRNA scaffold is already selected. Less experienced users are advised to proceed with these default settings by clicking the "Submit a task" button. For advanced users, the parameters can be adjusted within their allowed ranges. If a value outside the permissible range is entered, an error message appears, and the "Submit a task" button is disabled until the issue is corrected. This ensures that only valid configurations can be submitted for processing. If users modify parameter values and wish to restore the defaults, they can click the "Restore defaults" button.
A table below presents all design parameters, detailing their names, measurement units, recommended values, allowed ranges, and short descriptions. These descriptions can also be viewed directly on the Design parameterization page by clicking the "?" icon next to each parameter name.
| Parameter | Unit | Default value | Allowed values | Description |
|---|---|---|---|---|
| Min GC content | % | 30 | 0-100 | The minimum percentage of G and C nucleotides in the target sequence. |
| Max GC content | % | 65 | 0-100 | The maximum percentage of G and C nucleotides in the target sequence. |
| Max GC stretch | nt | 9 | 1-24 | The maximum allowed number of consecutive G and C nucleotides in the sequence. The presence of a G/C stretch longer than 9 nt should be avoided. |
| Bps to init binding | bp | 5 | 0-21 | The minimum number of base pairs required to initiate binding when checking off-targets. |
| Bps to init duplex dissociation | bp | 3 | 0-10 | The number of base pairs at both ends of the duplex that are checked to determine if dissociation starts from the correct site. |
| Min MFE difference between ends | kcal/mol | 2 | 0-100 | The minimum free energy difference between the dissociation energies of both duplex ends. One end must have a lower energy to dissociate first. |
| Max 2D structure variance | nt | 4 | 0-150 | The maximum allowed number of differences between the 2D structure of the designed amiRNA and the natural pre-miRNA scaffold. |
| Max melting temperature | °C | 21.5 | 0-100 | Maximum allowed melting temperature (Tm) for the duplex formed in the seed region. |
| Allow different prefixes | - | ON | ON/OFF | Specifies whether guide strands with prefixes different from the natural pre-miRNA scaffold should be included or excluded. |
| Filter off-targets | - | OFF | ON/OFF | Specifies whether off-targets should be considered or removed at every step of the design process. |
| Invalid prefix penalty | - | 2.5 | 0-5 | A penalty added to the amiRNA score if the prefix differs from the natural pre-miRNA scaffold. |
2.4. Modifying uploaded input data
Users can modify their input data at various stages of the specification process. On the Data upload page, they can change the input sequence, select a different gene/transcript identifier, or upload data from another file or example. On the Target specification (step 2) and Design parameterization (step 3) pages, they can return to previous steps by clicking the "Go back" button to adjust their target selection or design parameters before proceeding.
2.5. Running the program
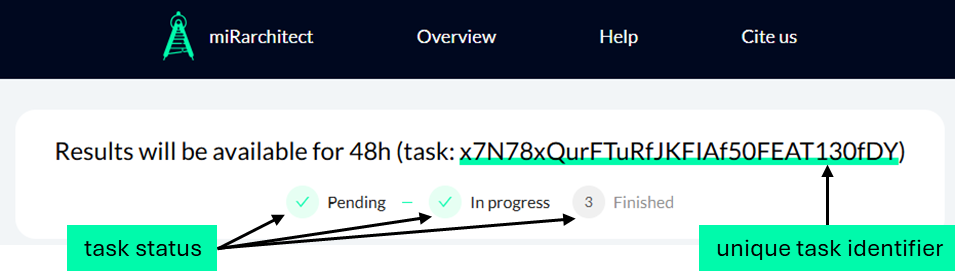
Users should click the "Submit a task" button on Design parameterization page to initiate data processing. This triggers the miRarchitect engine and displays a processing page indicating that amiRNA design is in progress. At the moment the user starts the processing, the task is assigned a unique 32-character identifier (TID). A dynamic loading animation informs users that computations are underway, which may take some time. Users can return to other activities and copy their unique task identifier, displayed at the top of the page. Left-clicking the identifier allows automatic copying to the clipboard. Users can later visit the results page and retrieve their designed amiRNA sequences using this TID.
Preview
3. miRarchitect results
The Results page appears immediately after the computations are completed and is accessible via a URL that includes the unique task identifier. The results remain available in the system for 48 hours after processing is finished. miRarchitect designs candidate amiRNAs, provides their characteristics, and reports detected off-targets. This information is organized into several sections on the results page, which are described in the following paragraphs.
3.1. Effective amiRNA candidates
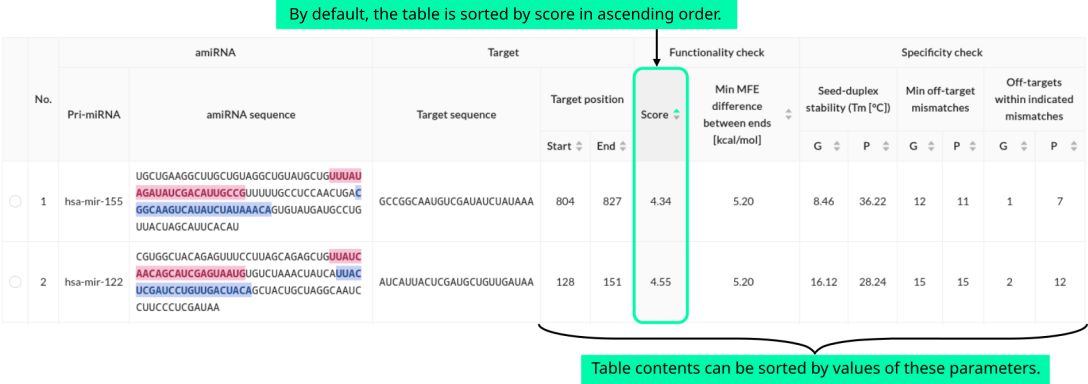
The main result generated by miRarchitect is a list of designed amiRNA candidates. The candidates are presented in a table, prioritized by a score reflecting their predicted functionality (lower scores indicate higher effectiveness), along with several parameters. For each amiRNA candidate, the following information is provided: candidate number, pri-miRNA scaffold, amiRNA sequence (with guide and passenger strands color-coded for easy identification), target sequence, target position (start and end), overall score, minimum MFE difference between the ends, seed-duplex stability (i.e., Tm for the guide and passenger strands separately), minimum number of off-target mismatches (separately for guide and passenger strands), and the number of off-targets within the specified mismatch thresholds (separately for guide and passenger strands).
The table can be sorted by any numerical value in ascending or descending order by clicking the arrows in the respective column headers. Clicking the radio button in the first column allows users to view more detailed information about a specific amiRNA candidate, which is displayed below the off-target statistics.
Preview
If the table contains more than ten candidates, it will be displayed in subtables with 10 entries per page. The table may require scrolling to the right to view all columns. Additionally, the entire table can be downloaded and saved as a CSV file.
3.2. Off-target statistics
The next important result is the off-target statistics. For each amiRNA candidate in the first table, users can see whether any off-targets were detected for that candidate, separately for the guide and passenger strands, across different regions: CDS, 5′UTR, 3′UTR, and ncRNAs. These data allow users to filter out candidates with a high number of off-targets in specific regions, such as the 5′UTR, or to select candidates with no off-targets for further studies. Off-target statistics are provided for mismatches ranging from 0-5. A dash ("-") in a table cell indicates no off-targets meeting the criteria, while a positive number indicates the number of off-targets in a specific region with the specified number of mismatches.
When a candidate is selected in the amiRNA candidate list, the corresponding row in the off-target statistics table is highlighted.
If the table exceeds ten rows, it will be split into subtables with 10 entries per page.
3.3. Annotated input sequence
The next section of the Results page is displayed upon selecting an amiRNA candidate from the first table. This section shows the full input sequence, with the target (complementary to the guide strand) and any detected off-targets highlighted.

Preview
3.4. amiRNA structural stability indicators
amiRNA structural stability indicators are displayed when users select an amiRNA candidate from the first table. The data in this section are computed by comparing the selected amiRNA candidate with the natural pri-miRNA scaffold. Users can view the sequence of the amiRNA candidate, a heatmap with color-coded Δ Shannon entropy values, and color-coded Δ free energy values. Below the heatmap, the secondary structure of the amiRNA candidate is shown in dot-bracket notation. Differences in the 2D structure relative to the pri-miRNA are highlighted with a grey background. Each column of the heatmap corresponds to a nucleotide position in the displayed sequence. Users can narrow the heatmap view to a specific sequence region using sliders located above the sequence. When hovering over a cell in the heatmap, the exact parameter value and the corresponding nucleotide symbol are displayed.
The heatmap can be downloaded and saved as an SVG file by clicking the "Download" button.

Preview
3.5. Off-target candidates for the guide/passenger strands
The final section of the Results page presents information about off-targets for the selected amiRNA, which is chosen from the list of effective amiRNA candidates displayed at the top of the page. The data is organized in a table that shows off-targets with a perfect match and those with up to 5 mismatches (≤17 complementary nucleotides) in either the guide or passenger strand. This result is based on a search for off-targets in 22-nt sequences from both strands of the siRNA duplex. The table includes the following data: accession ID, strand type (sense/antisense), number of mismatches, alignment (to help identify mismatch locations), region where off-targets were detected (CDS, 5′UTR, 3′UTR, or ncRNAs), and a description. The contents of the "description" column can be searched for specific keywords. To do so, users should click the magnifying glass icon placed in the header of the description column, enter the keyword, and click the "Seach" button. Clicking the "Reset" button in the pop-up window restores the full table view.
The table is sorted in ascending order by the number of mismatches by default, but it can also be sorted by strand type, number of mismatches, and region. If no off-targets with 0--5 mismatches are detected for the selected amiRNA, the table will not be displayed. If the table exceeds ten rows, it will be split into subtables with 10 entries per page. The table can be also downloaded and saved as an CSV file by clicking the "Download" button.

Preview
3.6. Returning to the result page
If a user has started a task and saved/copied its identifier, the Results page can be accessed from any device and browser within 48 hours after task processing is completed. To do this, users should go to the miRarchitect home page (which also serves as the Upload data page), and in the last section: (4) Provide a task ID (to retrieve previous results), enter the task identifier and click the "Get results" button. If the identifier is correct and the results are still available in the system, the Results page will be displayed.

Preview
4. System requirements
miRarchitect is designed to work with most of the available web browsers. The latest versions of browsers are strongly recommended.
| Operating system | Recommended browsers |
|---|---|
| Windows | Mozilla Firefox (134.0 and later), Opera (117.0 and later) and Google Chrome (135.0 and later). |
| Linux | Mozilla Firefox (134.0 and later), Opera (117.0 and later) and Google Chrome (135.0 and later). |
| MacOs | Mozilla Firefox (134.0 and later), Opera (117.0 and later) and Google Chrome (135.0 and later). |
5. Licenses
Researchers from academic institutions are eligible to use miRarchitect freely for academic purposes. However, any commercial use of the system requires a licensing agreement. To inquire about licensing, please contact marta.olejniczak@ibch.poznan.pl.